A very tough eval for protein language models
Two things that I believe to be true, based on my experience. First: the field of ML advances by evals—tasks and datasets. New algorithms chase evals and applications. Second: we are just starting to build really good evals for protein language models.
I believe that we can advance the field of ML protein design by creating new, tough evals for our models, examining the weaknesses in the predictions made by those models, and then dreaming up new models to beat the state of the art on the eval.
This post introduces the Bagel eval, a tough eval for protein design that's focused on the nuances of predicting enzyme function in a relevant protein engineering scenario.
A very tough eval for protein design
To do this, we'll use a model system created during my PhD where my coauthors and I experimentally measured kinetic constants for hundreds of mutants of a glucosidase enzyme and then also experimentally measured functional melting curves for the same set of mutants. This dataset and model system have a number of important characteristics that shape this eval. Overall, the dataset consists of kinetic constants for over 100 designed variants of BglB, a bacterial β-glucosidase with a solved crystal structure including a transition-state inhibitor catalyzing the hydrolysis of p-nitrophenyl-β-D-glucoside (pNPG), which is itself colorless. pNPG is a molecule of glucose linked to the colorful molecule p-nitrophenol, and when the enzyme hydrolyzes the bond, the p-nitrophenol released is bright yellow. The experimental measurements in the dataset span seven orders of magnitude, and represent a ton of careful work by a large team.
Like 99% of real-world protein engineering challenges, the desired substrate we are designing for is not the "natural" substrate for the enzyme (meaning that it is not the substrate the enzyme evolved to bind). It is almost always the case that this is true in a real-world protein engineering project. It's not the fastest enzyme in the world, or the slowest. It's easily expressed as a soluble monomer in E. coli. And the experimental assay can be performed with equipment you can find in any bio lab: just a plate reader, various kinds of plates, and multichannel pipet.
But the most important part of the eval is that we have experimentally measured both and for each enzyme, as well as , meaning that we know for each enzyme not only how good of a catalyst it is, but also how tightly it binds to the substrate. In practical protein engineering, both are fundamentally important to achieving the desired performance envelope.
Any model that we are using to design enzyme function should be able to predict the effects of mutation on catalysis and binding in this simple system. Right?
The new Bagel eval
We use this dataset to construct a difficult evaluation procedure for generative ML approaches to protein design, the Bagel eval, that we can use to evaluate generative ML approaches for protein engineering in a way that is complementary to existing benchmarks.
Design eval
The design eval consists of
- Spearman rank correlation between model prediction and
target=kcat - Spearman rank correlation between model prediction and
target=km
Evaluation walkthrough with ESM-2 and Progen2
Results summary:
| Method | Target | Spearman |
|---|---|---|
| ESM-2 (150 M) | kcat | 0.58 |
| ESM-2 (150 M) | km | –0.24 |
| Progen2 (151 M) | kcat | 0.59 |
| Progen2 (151 M) | km | –0.19 |
| Rosetta + ElasticNet | kcat | 0.57 |
| Rosetta + ElasticNet | km | 0.68 |
While protein language models are able to match structural features and regularized linear models on the kcat target, they perform very poorly on the km target when compared to a structural ML approach.
Process the mutant dataset
Find the mutant dataset in data/dataset.csv. This contains the single-mutant data for the BglB enzyme. Note the two columns "kcat" and "km" which are the log-transformed targets. For each sequence, we hope to be able to predict the kcat (catalytic rate) and km Michaelis constant, which quantitates substrate affinity.
Predict functional effects of single variants
To predict the functional effects of mutations using a generative model, there are a few different ways to think about it. One good discussion of the merits of the various approaches can be found in "Language models enable zero-shot prediction of the effects of mutations on protein function", a paper from Alex Rives' group (the group that developed ESM). In the paper, Meier examines various different ways of scoring mutations with language models. We will take the method that is most accurate in their testing (masked marginals), but also note that there is a method that is both efficient and accurate, WT marginals. To calculate this, "[p]robabilities are extracted according to the mask noise during pre-training. At each position, we introduce a mask token and record the model’s predicted probabilities of the tokens at that position" (from the first page of the supplemental material).
Using ESM-2
Use the provided script predict.py to predict the effects of mutations using ESM-2 using the masked marginals approach. I selected a small ESM-2 model (150 M params to match the smallest Progen model, see below).
python predict/predict.py \
--model-location esm2_t30_150M_UR50D \
--sequence "MSENT (...snip...) KNGF" \
--dms-input data/dataset.csv \
--offset-idx 1 \
--dms-output data/dataset_labeled_1.csv \
--scoring-strategy masked-marginals
This will provide zero-shot predictions from ESM-2 for the BglB dataset.
Using Progen2
Use the provided script from ProteinGym with slightly modified input options (available in models/progen2/predict.py) to predict the effects of mutations using Progen2. We'll use the smallest Progen2 model, progen2-small with 151 M parameters.
python models/progen2/predict.py \
--model progen2-small \
--sequence "MSENT (...snip...) KNGF" \
--dms-csv data/dataset_labeled_1.csv \
--output-csv data/dataset_labeled_2.csv
Evaluating the zero-shot predictions
Before we evaluate the performance, let's first think a sec about what we're asking for here. We are going to calculate the effect of a mutation by sending in the sequence to the model, retrieving the logits until we're looking at the logits for the mutant residue. We are looking directly at the probability distribution over possible tokens when the model comes to look at the position we are mutating.
It's worth thinking broadly about our problem setting as well. Here, we have two targets. One target is , which is the number of substrate molecules the enzyme "turns over" per unit time. In this case, the enzyme is hydrolyzing a glycosidic bond. The substrate is a synthetic reporter consisting of a sugar molecule linked to a molecule that becomes colored on release. In nature, there exists no selective pressure for the enzyme to efficiently work on this substrate, and this is reflected in the kinetic parameters for the wild type enzyme when acting on pNPG.
What we have is a classic protein engineering case study. We have some desired chemical transformation. In our case, the hydrolysis of glucose-pNP to glucose and pNP. We have found some natural enzyme that works, sort of, and does our reaction. But we would like to increase the substrate affinity by mutating the enzyme and observing the results in a biochemical assay.
Now consider the training datasets for models like ESM and Progen. In the case of these large transformer models, the training data consists of large datasets of natural protein sequences. Of which our WT sequence is actually one. These models are trained to recover a masked token, or predict the next token, during training.
Let's consider: do we expect a protein language model to be able to predict catalysis and binding, when it doesn't model small molecules at all, and is provided no information about what reaction we're trying to catalyze or the structure of the molecule (or of the protein, for that matter). It's a very, very difficult challenge and we might expect very poor performance.
OK, so that out of the way, we can use the notebook evaluate.ipynb to draw plots and look at predictions.
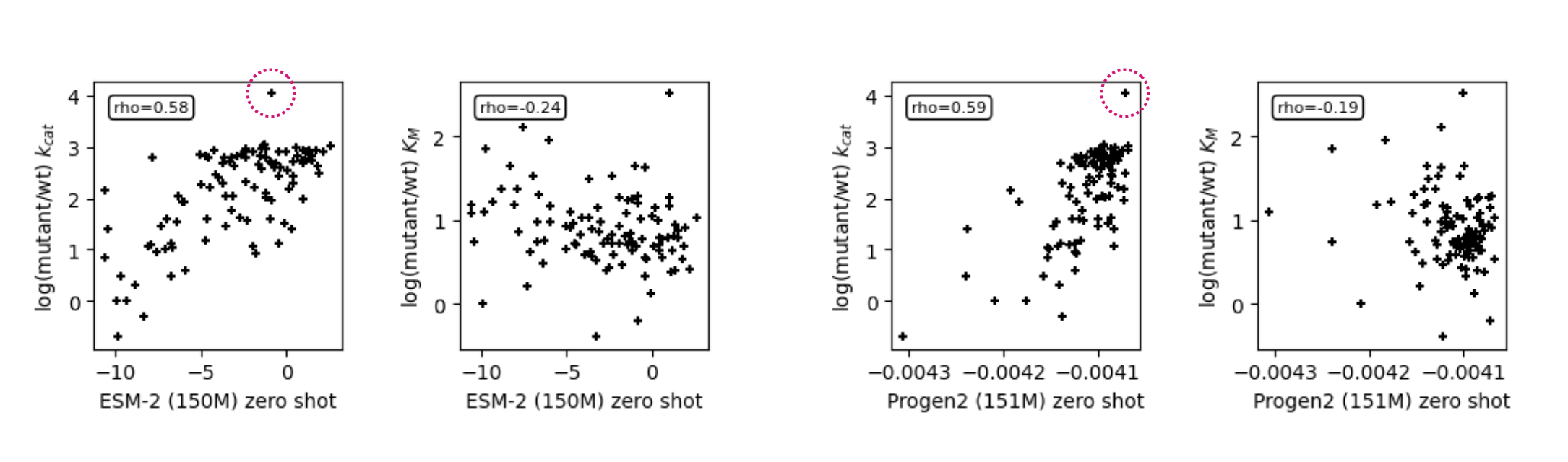
These scatter plots showw predictions by models from the ESM-2 (left two plots) and Progen2 (right two plots) families of around 150 M parameters for kinetic constants and . In the plots for , I have circled the datapoint with the highest experimental value. Note now Progen correctly predicts both the highest and lowest correctly.
The predictions for are actually ... pretty good!
It is fantastic to see that these large protein language models both capture the deleterious effects of mutations on the catalytic rate. Both the "encoder-only" ESM model trained with a masked language modeling objective and the "decoder-only" Progen2 model trained via next token prediction are able to achieve a Spearman rank correlation of near 0.60.
I don't want to put too much stock in a single datapoint, but I think it's worth noting—and I've marked it on the graphs—that while ESM incorrectly predicts the mutant with the highest as about average, Progen2 correctly predicts the value for this mutant as among the highest in the dataset. Progen2 also correctly places the lowest datapoint (same as ESM).
Compared to ESM, Progen also scores better on both common measures of correlation for regression problems (Spearman and Pearson) for the catalytic rate target.
Conceptually, I think it makes sense that large protein language models can predict deleterious effects of mutations. Progen's ability to correctly predict a mutant that is better than the wildtype implies that it may be a better model than ESM for design.
No correlation with predictions for
This I think is the interesting bit. In our paper, we showed that by modeling the enzyme–transition state complex for each of the mutants in the dataset using molecular mechanics (Rosetta), calculating structural features from the models, and then using those features as the input to a regularized linear model (Elastic Net), we could predict both the kcat and km targets with Spearman rank correlation of 0.57 for kcat and 0.68 for km.
While the protein language models here capture the deleterious effects of mutations on the enzyme's , there is no information provided to the model that would allow it to estimate the binding constant for an arbitrary substrate, in this case, pNPG. In building the enzyme–transition state complex structure for all the mutants, we are directly providing as much as we can about the system. The language models receive only the sequence and therefore have no basis for predicting a binding constant.
Conclusions
This post introduced the Bagel eval, a tough eval for protein design that's focused on the nuances of predicting enzyme function in a relevant protein engineering scenario. Using a high-dynamic range dataset of kinetic constants and for over 100 variants of BglB, we explored the ability of protein language models to predict the effects of mutations on these functional parameters.
Protein language models trained on sequences alone rivaled or outperformed a method involving all-atom molecular mechanics simulations of the enzyme–transition state when predicting the mostly-deleterious effects of mutations in this eval. In contrast to ESM, Progen was able to correctly rank the highest and lowest points in our dataset in this zero-shot setting for the kcat target.
For the km target, none of the protein language models examined showed any correlation with the experimental values, whereas the method in which we use molecular mechanics to simulate the entire system and use constrained statistical learning to make predictions achieved a SRC of 0.68 this dataset.
The Bagel eval points to exciting new avenues for improving algorithms for ML protein design, and provides a measurable means to track progress. Along with extensively documented wet lab protocols to expand the dataset, which can easily be applied to new proteins, we hope that the Bagel eval is a useful tool for improving the ability of protein language models to design functional proteins.