Fun with molecular transformers
I was curious to see if I could learn some useful embeddings of small molecules using language modeling, and perhaps generate some wacky new molecules as well. Since molecules can easily be represented using SMILES strings, it seems like a straightforward application of a character-level transformer model.
Picking a dataset
Our goal is to create a chemical dataset. We would like to include a wide variety of chemical species, and we would like to focus on those with biological relevance. This means roughly, that the molecules should be involved in biochemical reactions, bind to proteins, and be important, somehow, in biology.
The dataset must also be of a certain size and "shape", if you will. In this case, I'd like to find something with perhaps tens of thousands of examples, each of which is relatively "short" by language modeling standards. This small block size will allow us to be computationally efficient, while the number of samples should provide enough to adequately train a small language model.
In order to meet these criteria, I decided to use small molecules that are present in solved structures in the Protein Data Bank. Using PubChem, I retrieved metadata for 36,249 small molecules that appear in PDB entries. After examining the distribution of molecular weights, I filtered this down to a list of 33,883 small molecules, each of which appears in the PDB and has a molecular weight of between 30 and 650 g/mol.
Everything in the training set is represented as a SMILES string, a chemical formatting string that completely specifies heavy atoms and connectivity, including bond order and cycles. So examples range from hydroxylamine (molecular weight 33 g/mol) represented H3NO, to 9-ethyladenine, represented CCN1C=NC2=C(N=CN=C21)N, to N-(4-fluorophenyl)-4-methyl-1-piperazinecarboxamide (237 g/mol), represented CN1CCN(CC1)C(=O)NC2=CC=C(C=C2)F, to even larger molecules whose names and SMILES strings will both overflow the text box on this web page.
After tokenization, we have a vocabulary of 57 characters, with the longest individual sequence being 113 characters. Since we'll add a start token, this will make our model's block size 114. The dataset is shuffled and split into 80% training examples, 10% validation, and 10% test examples. (Perhaps we could partition the dataset in a different way, perhaps after clustering by chemical similarity, in another set of experiments.)
Which model to use?
Chemicals can be represented as graphs, where each atom is a node (vertex), and each bond is an edge. The use of graph neural networks to model chemical structures is well-established. However, for this project, I have something else in mind down the road, and well basically I would like to see if a transformer model is able to learn to generate new molecules in an autoregressive fashion.
In this case, we'll use a "decoder-only" transformer model, with 4 heads, 4 transformer layers with causal self-attention, a model dimension of 64, a block size of 114, and a batch size of 32 examples. This model is about 200k parameters. The number of tokens in our dataset is 1,606,412, so this is about 8 tokens per parameter if it's helpful to think of it in that way. The implementation is straight from Andrej Karpathy's excellent makemore series, which, along with being a nice, straightforward implementation, is a pedagogically fresh take on teaching language modeling that I personally find very elegant and refreshing.
We train this model for 60k steps, to a training cross entropy loss of 0.52 and a test loss of 0.64, which takes less than an hour on my M2 MacBook Air. The test loss seems to reach a minimum around 40k steps, and sneaks up a little bit right around 55k steps, so we select the model with the lowest test loss, which occurs around step 47k.
What kinds of molecules does the model generate?
To sample from the model, we use a temperature of 1.0 and generate 50 samples. This is a nice small number we can take a good look at.
For each molecule, I used RDKit to draw it
smiles = "CN1C=NC=C1"
fig = Draw.MolToImage(Chem.MolFromSmiles(smiles))
fig.save(format="png", fp="filename.png")
I found that of 50 molecules generated, 7 were invalid (largely due to unclosed rings), 3 were in the training set, and 40 were "valid" molecules that did not exist in the training set.
So naturally I picked a few of my favorites.
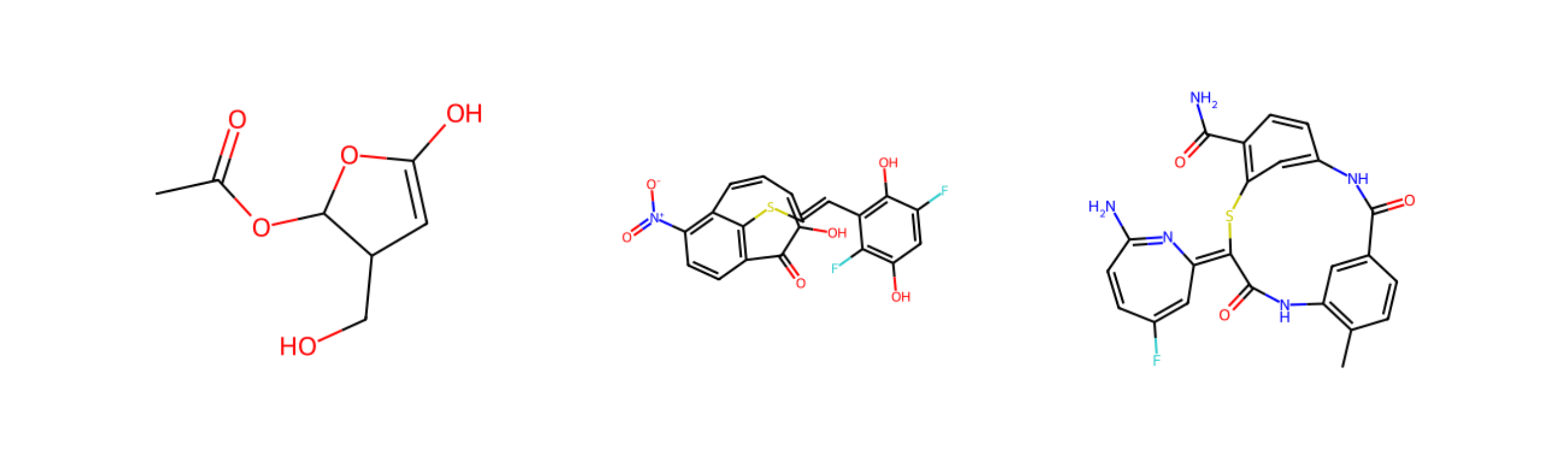
These generated molecules are, from left to right:
CC(=O)OC1C(C=C(O1)O)COC1=CC(=C2C(=C1C(=O)C(=CC=C2)O)SC=CC3=C(C(=CC(=C3F)O)F)O)[N+](=O)[O-]CC1=C(C=C(C=C1)C(=O)NC2=CC(=C(C=C2)C(=O)N)SC3=C4C=C(C=CC(=N4)N)F)NC3=O
Now, these are strange, don't appear in PubChem, and let's be honest, look wildly unstable. The fact that they are parsable SMILES strings doesn't mean they'll be nice stable molecules. These are some of the wildest ones, many look relatively reasonable.
However, the molecules generated by the network do have some interesting properties. They are in some sense freed from the constraint of being biocatalytically "reachable" in the known chemical history. They are not constrained to be made from chemical motifs created by the set of biosynthetic enzymes. If perhaps some of them were stable, it would probably be a challenge to synthesize them, as they are not rearrangements of existing biosynthetic motifs.
Can we improve upon this approach by using a model that better captures the structure of molecules? I have no doubt that using a graph-structured neural network could be used to improve upon this approach and generate better, more realistic molecules.
However, I have something slightly different in mind. I think it would be very interesting to train a unsupervised sequence model on pairs of {protein, ligand} where both the protein and the ligand are represented as token sequences. There is a ton of prior work showing that proteins can be represented by language models, and we have just seen how language models can represent the small molecule ligands in the PDB. The only thing missing is to put these together.