Multistate protein design with AlphaFold and ProteinMPNN
The central dogma of biology states that information flows from DNA to RNA to protein. In the field of protein design, we have a similar dogma, which is: sequence determines structure, and structure determines function. Proteins are linear polymers of amino acids that fold into complex 3D shapes to do chemistry. Thus, it's often highly desirable to have a structure for a protein that you seek to design. That's why AlphaFold is such a big deal.
However, one of the major shortcomings of structure-based design approaches is that they invariably use a static picture of a protein structure as input. However, we know that proteins are highly dynamic molecules in solution. For some proteins, like kinesin motion is what they do, and enzymes like ATP synthase are extraordinary molecular machines that spin like a propeller motor.
Some folks in the community believe that all enzymes benefit in some way from macromolecular motion. In my experience, there are many enzyme classes where macromolecular motion is an important determinant of catalysis. Mutations to the active site "lid" of terpene synthase enzymes render these enzymes unable to catalyze the desired reaction due to the failure of the lid to exclude water from the active site. The huge and diverse class of CoA ligase enzymes, which attach diverse molecules to the molecular "handle" known as coenzyme A, rely on large conformational changes that occur between successive steps of the enzyme mechanism.
So why do structure-prediction algorithms like AlphaFold and sequence-design algorithms like ProteinMPNN ignore all protein motion and deal with only static structures?
I hypothesized that a model that explicitly took into account different structural states would produce better and more highly-functional designs than a model that used static pictures alone. I found that training an ensemble of ProteinMPNN models that each accepted a different, energetically-favorable conformational state, then averaged logits during autoregressive decoding, produced a model that was forced to design sequences that matched all conformational states, not just the single one provided in previous approaches.
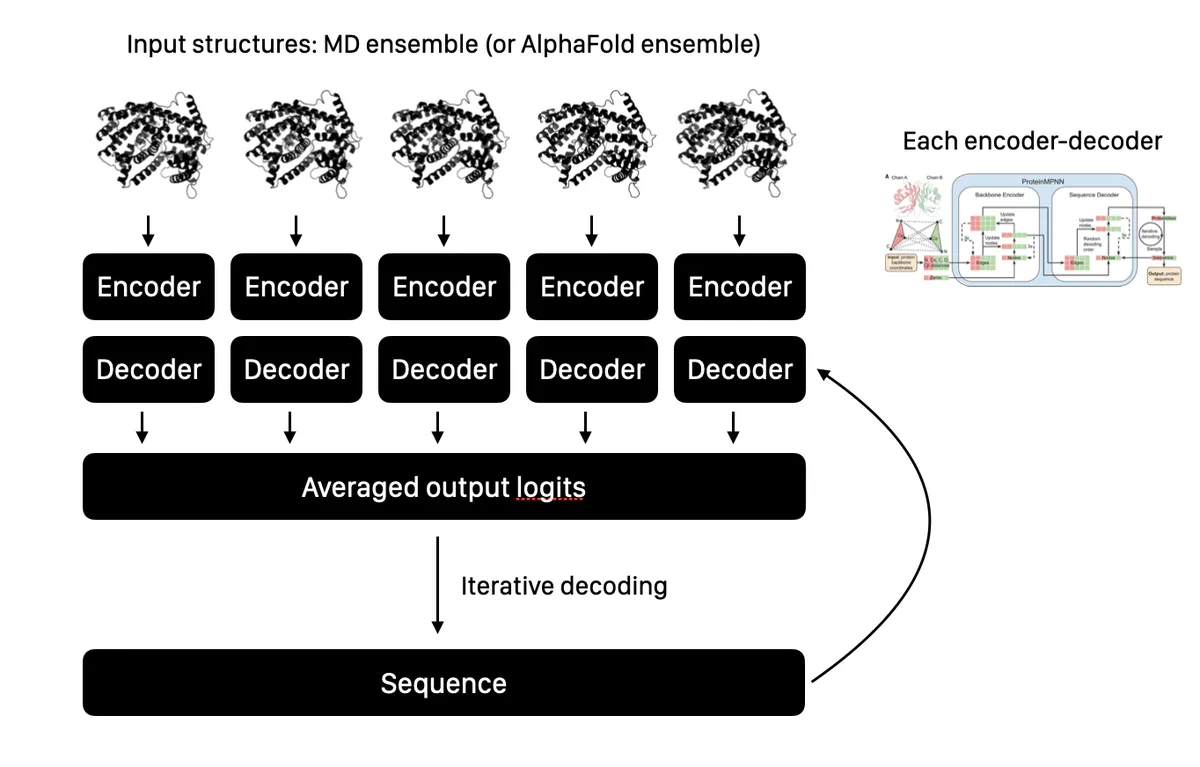
It turns out that using the ranked ensemble 5 models output by AlphaFold 2 works almost as well as the much more computationally–expensive technique of molecular dynamics. So one fun and interesting use of AlphaFold and ProteinMPNN is to build a model that models the different conformational states of your protein and can design new sequences that are compatible with them all. Why is this useful? This model tends to be much more conservative about destroying functional sites, while retaining the ability to design better expression and solubility. If the function of important proteins depends on more than a static picture, we need approaches that can model this motion to make better predictions.